
EUDAMED es la plataforma informática diseñada para recopilar y gestionar información sobre dispositivos médicos comercializados en Europa. Surge del reglamento EU 2017/745 (MDR) Y 2017/746 (IVDR) con el objetivo de garantizar la trazabilidad de productos sanitarios, consiguiendo una visión global sobre el ciclo de vida del producto. El objetivo principal de esta base de datos es la transparencia para todo el profesional sanitario y reforzar la coordinación entre los Estados miembros de la UE, es decir, mejorar acceso a la información a fabricantes, organismos notificados y autoridades competentes.
Proceso de Adaptación y tiempos para la implementación de los módulos obligatorios
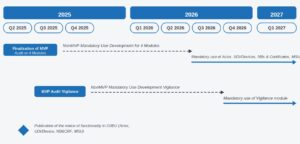
¿Qué estructura tiene?
La base de datos cuenta con seis módulos, los 4 primeros, obligatorios desde mayo 2026.
- Módulo 1: Registro de actores: Incluye fabricantes, representantes autorizados e importadores. Punto de entrada a EUDAMED donde todo operador económico tiene que registrarse. El registro implica que deben tener el SRN (SingleRegistration Number), es decir, un identificador único que permite a las autoridades reconocer a cada actor dentro del sistema informático. Esto implica mayor control, obligación de mantener los datos actualizados y operar plenamente bajo MDR/IVDR. Para considerar registrado a un actor económico es imprescindible dos premisas:
- Disponer de solicitud de registro de actor validada
- Disponer de un ID de actor/SRN: La identificación del actor, es decir el ID/SRN será la siguiente:
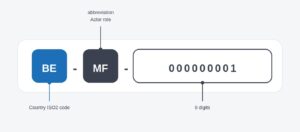
Como Actores tenemos:
- Fabricantes: Deben registrarse, gestionar con sus representantes autorizados, registrar sus productos y los informes correspondientes de vigilancia y control poscomercialización.
- Importadores: deben registrarse, asociarse a los fabricantes no pertenecientes a la UE cuyos productos comercializan y pueden consultar los datos de dichos fabricantes y productos introducidos en EUDAMED
- Representantes autorizados (RA): se registran, participan en el registro del fabricante no perteneciente a la UE, participan en la gestión de los mandatos, registran los informes de vigilancia cuando procede y verifican la
información facilitada por los fabricantes no pertenecientes a la UE.
- Módulo 2: Registro de dispositivos (UDI): Contiene información detallada sobre los productos, incluyendo el Identificador Único de Dispositivo (UDI). Es un módulo complejo, en realidad se trata del más crítico ya que se centra en la identificación del dispositivo sanitario. El UDI facilita datos tipo: Información del fabricante o incluso datos básicos del dispositivo esenciales para su trazabilidad. El UDI está compuesto por dos partes, UDI-DI (Identificador del dispositivo) + UDI-PI (Identificador de producción). Indispensable para la integración con sistemas internos ERP de la propia empresa.
- Módulo 3: Certificados y organismos notificados: Recoge los certificados emitidos y la información sobre los organismos que los han evaluado. Módulo que se crea para facilitar la gestión de los certificados de los dispositivos sanitarios a los fabricantes. Se trata de un módulo que incluye certificado CE, alcance del certificado y estado de vigencia. Esto implica mayor transparencia sobre la validez del certificado, mayor acceso a las autoridades y dispositivos en el mercado certificados y validados. En resumen, es un módulo que facilita la verificación regulatoria reduciendo el riesgo de falsificaciones o incluso certificados inválidos o desactualizados.
La información de los organismos notificados se encuentra en la base de datos NANDO
- Módulo 4: Vigilancia y post-comercialización: Permite reportar incidentes graves y acciones correctivas de seguridad. Es un módulo centrado en los dispositivos puestos en el mercado. Importante para conocer notificación de incidentes graves. Es un módulo dirigido especialmente a las autoridades competentes, ya que en lo que respecta a la vigilancia del mercado, la mayoría de las funciones y obligaciones les corresponden a dichas autoridades.
- Módulo 5: Investigaciones clínicas y estudios de funcionamiento: Registra estudios clínicos relacionados con los dispositivos. Es un módulo que incluye solicitudes de autorización, protocolos y resultados finales. Se ha creado para conseguir mayor armonización a nivel europeo, transparencia de todos los ensayos clínicos e interacción entre autoridad y comité ético.
- Módulo 6: Supervisión del mercado: Facilita la cooperación entre autoridades para el control del mercado. Módulo que refuerza e incrementa el control del dispositivo en el mercado.
Impacto dentro de la organización
La introducción de EUDAMED supone un alto impacto en el sector, implicando a fabricantes, importadores y distribuidores. En ocasiones se ve como una carga extra en el sector ya que:
- La empresa debe asegurarse estar correctamente registrada y actualizar continuamente los datos.
- Implementación de un sistema ERP para mantener la gestión del UDI y por lo tanto la trazabilidad del dispositivo.
- Mayor carga documental, más información regulatoria con más detalle y más información técnica.
- Adaptación de protocolos internos, desde la parte de calidad hasta la parte IT de la empresa que se ven implicadas en este proceso.
Retos y oportunidades
EUDAMED constituye un cambio de mentalidad para muchas empresas,
- Mayor confianza en el producto puesto en el mercado: A través de EUDAMED se ofrece el acceso a información actualizada de todo tipo de producto sanitario lo cual genera un nivel mayor de TRANSPARENCIA para el profesional sanitario (hospitales, distribuidores…) gracias a que, en todo momento se podrá verificar fácilmente el estatus regulatorio de un producto, tomando decisiones rápidas y firmes, en paralelo, ofrece mayores GARANTÍAS de seguridad frente al paciente,
- Productos más visibles: Gracias al volcado de datos de todas las empresas, EUDAMED como base de datos ofrece en todo momento una información regulatoria que puede ser consultada en todo momento
- Optimizar procesos: Centralización de datos, estandarización y automatización además de mayor trazabilidad.
¿Qué ha cambiado desde el 28 de mayo de 2026?
Desde esta fecha marcada ya en el calendario:
- EUDAMED pasa de ser opcional a ser obligatorio para todos los operadores económicos
- Los 4 primero módulos serán de uso obligatorio:
- Registro de actores
- Registro UDI
- Certificados y ON
- Supervisión en el mercado.
Esto significa que no se podrá comercializar nuevos dispositivos dentro de la Unión Europea sin estar previamente registrados en EUDAMED, es decir, se convierte en un requisito de acceso al mercado.
Sí que es cierto que, existe un periodo de transición “Legacy devices” que hace más cómoda la adaptación, dando tiempo a las empresas hasta el 27/28 de noviembre para registrar todos sus dispositivos.
Conclusión
EUDAMED se convierte en poco tiempo en el eje central dentro del modelo europeo de productos sanitarios construyendo así transparencia, conexión y exigencia. Con la entrada en vigor de la obligatoriedad, el sistema se convierte en un requisito crítico de acceso al mercado.
Este cambio no solo impacta en el cumplimiento normativo si no que constituye un proceso de transformación para las empresas ya que requiere de una organización de su información, procesos y estrategia regulatoria.
En este sentido, EUDAMED da el paso a una mayor confianza del dispositivo médico en el mercado no solo para el profesional sanitario si no para el paciente.

