
La Comisión Europea ha lanzado el borrador del Reglamento Delegado (UE) 2024/1701 (UE) por el que se modifica el Reglamento (CE) n.o 1234/2008, en lo que se refiere a las modificaciones de los términos de las autorizaciones de comercialización (AC) de medicamentos para uso humano (Reglamento sobre variaciones) dentro del marco jurídico vigente del Reglamento (CE) nº 726/2004 y la Directiva 2001/83/CE.
Periodo de transición y objetivos del nuevo reglamento
Si bien, el 1 de enero del 2025 entró en vigor un primer borrador del reglamento delegado, con el principal objetivo de conseguir un marco jurídico más simple, claro y flexible, así como garantizar el mismo nivel de protección de la salud pública, a día de hoy nos encontramos en un periodo de transición, a la espera de una implementación definitiva a partir del 15 de enero de 2026 cuando salgan las publicaciones oficiales nacionales de un segundo borrador que incluye aclaraciones y nuevos conceptos sobre el vigente.
Estas actualizaciones de la llamada “guia de variaciones” de medicamentos de uso humano pretende, entre otras cosas, reducir la carga administrativa tanto para las autoridades como para los Titulares de Autorización de Comercialización (TAC).
En definitiva, nos encontramos ante otro avance en la reforma legislativa para crear una mayor Armonización entre todos los Estados Miembros, así como para optimizar los recursos.

El Reglamento Delegado 2024/1701, propone cambios en la clasificación de las variaciones y cambios sobre la aplicación de los procedimientos establecidos en diferentes capítulos de la misma. La mayor parte de los cambios en la clasificación se debe fundamentalmente a los nuevos requerimientos introducidos por la legislación de Farmacovigilancia de la UE y otros cambios en Calidad.
En el marco introductorio, se mantienen las alternativas de las ampliaciones en las autorizaciones de comercialización (Extensiones de linea), junto con el enfoque prioritario hacia la protección de la salud pública mediante las actualizaciones de las vacunas frente a la gripe y el coronavirus (Annual Updates). Además, se enfatizan la importancia de implantar medidas regulatorias urgentes (Urgent Safety Restrictions, URS) ante situaciones de riesgo, asegurando la seguridad sanitaria.
¿Qué cambios se observan a lo largo del tiempo?
A partir del 1 de enero del 2025, las Autoridades publican los primeros cambios sobre la regulación de las variaciones, encaminados a facilitar trámites administrativos tanto a los laboratorios como a las propias agencias europeas que los tienen que evaluar, empezando por un objetivo claro: medicamentos de uso humano. En este contexto, aparece además una nueva versión del eAF (electronic application form) para implementar todos estos cambios propuestos.
Si bien es cierto que se mantiene la misma clasificación de variaciones que había hasta la fecha (Variaciones menores tipo IA, IB, y mayores Tipo II, extensiones de línea y restricciones de seguridad urgentes), en el 2025aparece, por primera vez, nuevos términos para variaciones menores tipo IA, como por ejemplo el de Super Grouping y Annual Report y se enfatiza el concepto de worksharing para variaciones TIB y TII.
Actualmente, disponemos de un nuevo borrador, que está en periodo de traducción y que se implementará a partir del 15 de enero del 2026; en él, no solo se mantienen todos los cambios propuestos en este periodo de transición vigente desde el 1 de enero del 2025 sino que además incluyen algunas aclaraciones en cuanto a procedimientos y gestiones administrativas, además de algunos cambios relevantes en los Anexos.
Aparecen nuevas aclaraciones en la introducción, tales como la incorporación de nuevas definiciones, que, en las versiones de años anteriores no estaban presentes. Se acuña el concepto de “Relevant Authority” (autoridad competente de cada miembro concernido o agencia en el caso de procedimientos centralizados) y “Reference Authority” (en el contexto de supergroupings y workshring, la agencia seleccionada para actuar como el referente de la evaluación de la modificación propuesta) para dirigirse a las diferentes autoridades que intervienen en el proceso de gestión y evaluación.
En el contexto de la gestión de las modificaciones a presentar (ya sean TIA, TIB o TII), se define el término “Grouping” que no estaba incorporado en el Reglamento (CE) n.o 1234/2008 como concepto, aunque se usaba siguiendo directrices europeas.
Adicionalmente, en relación a las variaciones menores TIA, el nuevo término acuñado en 2025 “supergrouping” se mantiene para este tipo de variaciones.
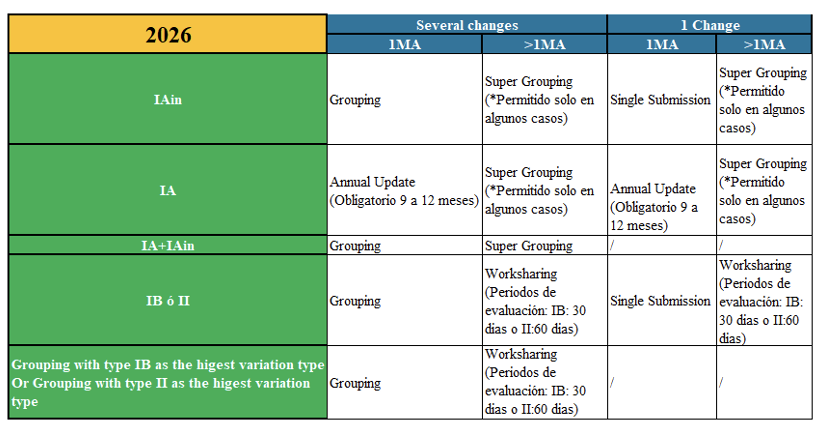
Supergrouping
El super grouping (grupo de modificaciones IA/IA-IN que puede presentarse en una única notificación para varias autorizaciones de comercialización (AC) propiedad del mismo titular, siempre que las modificaciones notificadas sean idénticas para todas las AC en cuestión. Requiere de confirmación previa por parte de las autoridades para su presentación, dado que es necesaria la asignación del número de variación), se hace posible en los siguientes casos:
- Una o varias modificaciones de importancia menor de tipo IA, notificadas al mismo tiempo para varias autorizaciones de comercialización concedidas con arreglo al procedimiento de reconocimiento mutuo, descentralizado o puramente nacional en varios Estados miembros.
- Una o varias modificaciones tipo IA notificadas al mismo tiempo para varias autorizaciones de comercialización concedidas con arreglo al procedimiento de reconocimiento mutuo o descentralizado y el Estado miembro de referencia es el mismo para dichos procedimientos.
- Una o varias modificaciones de importancia menor de tipo IA notificadas al mismo tiempo para varias autorizaciones de comercialización concedidas con arreglo al procedimiento centralizado.
Siguiendo con los cambios en las variaciones menores tipo IA, en el 2025, aparece el término “Annual Reporting”, lo que hace posible que variaciones tipo IA (ya sean TIA, TIA-IN y/o combinación de ambos) se recopilen y se presenten como un Annual Update, siempre que cumplan con las condiciones de la guia. Las variaciones TIA incluidas en esta agrupación no necesitan estar relacionadas entre sí. Se deberán presentar en el plazo de doce meses (de 9 a 12) tras la aplicación de la primera modificación. Este tipo de notificación no permite la mezcla de procedimientos: o todos nacionales o todos europeos, pero no se pueden mezclar entre ellos.
Refuerzo del worksharing
Adicionalmente, se refuerza el concepto worksharing para variaciones menores tipo IB así como para las variaciones mayores tipo II, cuando afecte a varias autorizaciones de comercialización (del mismo titular). El periodo de evaluación de la modificación mayor será el considerado (IB: 30 dias o II:60 dias). En versiones anteriores, se daba la opción al titular para uso, ahora se enfatiza su uso.
Considerando la legislación emergente de dispositivos médicos y su interrelación con producto farmacéutico, la guía de variaciones también muestra modificaciones relevantes en este sentido; es por ello que cuando un medicamento requiera del uso de un producto sanitario para su aplicación, se tendrá en cuenta la nueva regulación de dispositivos médicos y las nuevas guías de calidad documental para medicamentos, midiendo el impacto/riesgo en la administración, calidad, seguridad y/o la eficacia del medicamento. No podemos olvidar que, los productos combinados (compuesto por dos o más componentes regulados, es decir, fármaco/dispositivo, producto biológico/dispositivo, fármaco/producto biológico o fármaco/dispositivo/producto biológico, que se combinan o mezclan física, química o de cualquier otra forma y se producen como una sola entidad) tienen que tener una regulación estricta y controlada tanto del producto sanitario/dispositivo que se utilice como del fármaco que se aplique.
Reestructuración de los Anexos
En definitiva, la propuesta presentada y en uso de la guía de variaciones actual, implica una reestructuración de la misma con una visión mas simplista sobre la categorización de las variaciones.
Pero dentro de este documento “Draft” en el 2026, el gran cambio que afecta a los Anexos, es el cambio en la clasificación que se mantenía desde el 2024 que afecta a los 4 capítulos de los Anexos que hasta ahora se clasificaban como ABCD, a partir del 2026 serán EQCM:
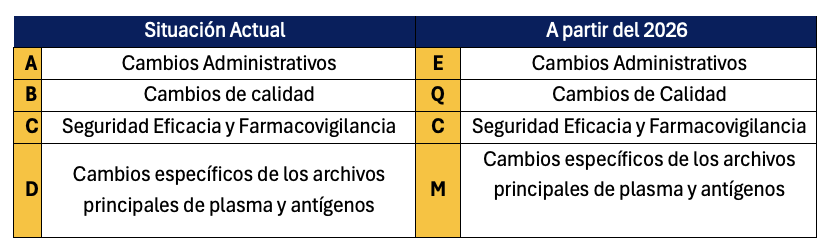
El reglamento Delegado 2024/1701 aporta a la industria farmacéutica un avance más en lo que respecta a la claridad, plazos más definidos y herramientas tipo Supergrouping y Annual Update tan necesarias para reducir los trámites administrativos y facilitar a los Titulares de Autorización de Comercialización (TAC) una gestión más optimizada, reforzando la transparencia en todos los procesos.

