
El procedimiento descentralizado (DCP) se utilizará para obtener autorizaciones de comercialización en varios Estados miembros cuando el medicamento en cuestión aún no haya recibido una autorización de comercialización en ningún Estado miembro en el momento de la solicitud.
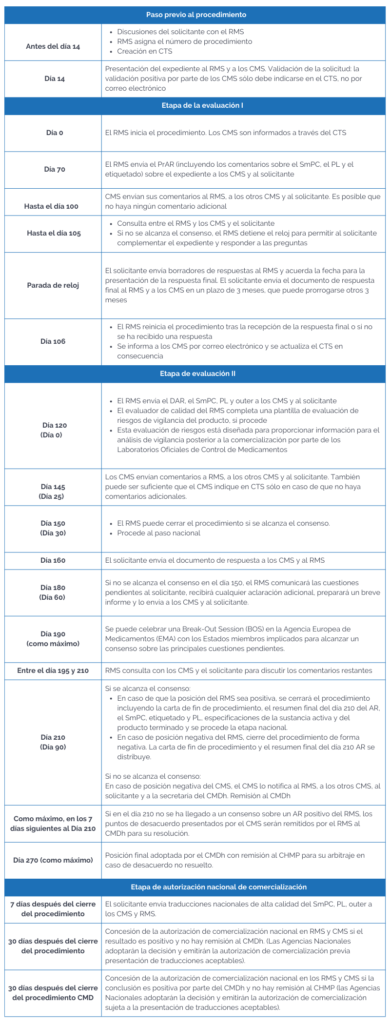
El DCP se divide en cinco etapas:
- Etapa previa al procedimiento, incluida la fase de validación
- Etapa de evaluación I
- Etapa de evaluación II
- Discusión a nivel de grupo de coordinación (CMDh), si es necesario
- Etapa de autorización nacional de comercialización
Paso previo al procedimiento, incluida la fase de validación:
Consulta con RMS
El solicitante informa al Estado miembro elegido como Estado miembro de referencia (RMS) para estimar una fecha y presentar la solicitud. El RMS asignará un número de procedimiento y lo creará en la base de datos del CTS para que los estados concernidos (CMS) y el RMS puedan comunicarse. El solicitante debe tener toda la documentación perteneciente al dossier y tener preparada su propuesta para la ficha técnica (SmPC), prospecto (PL) y etiquetado (outer).
Cómo presentar la solicitud
Se debe presentar una solicitud a cada país en el que se quiere comercializar el producto con una carta de presentación declarando que el dossier presentado es idéntico en todos los Estados miembros afectados (RMS + CMS).
Fase de validación
Tras la presentación de la solicitud, tanto el RMS como los CMS, deben informar al solicitante sobre cualquier cuestión administrativa y reglamentaria. Una vez finalizado este plazo y con la confirmación de todos los Estados miembros afectados, comenzará la evaluación del procedimiento.
Evaluación etapa I
El RMS envía el Informe de Evaluación Preliminar (PrAR) a los CMS y al solicitante en un plazo de 70 días tras el inicio del procedimiento.
Antes del día 100, los CMS deben comunicar sus comentarios al RMS y al solicitante, detallando las razones de su posición.
Entre el día 100 y 105, el RMS puede ponerse en contacto con los CMS para discutir los comentarios planteados.
En este punto puede suceder:
- Que el producto sea aprobable, en cuyo caso el RMS actualizará el PrAR y preparará el Informe de Evaluación Final (FAR). Una vez tramitado esto, se continuará con la etapa nacional.
- Que el producto sea aprobable, pero hay algunos comentarios que se pueden resolver fácilmente. Entonces, el RMS detiene el reloj y envía los comentarios el día 105 al solicitante. Una vez recibida la respuesta del solicitante, el RMS reinicia el reloj en el día 106 y actualizará el PrAR y preparará FAR. Puede llegar hasta el día 120.
- Si el producto no es aceptable y todavía hay cuestiones importantes por resolver, se detiene el reloj en el día 105. Se le envían al solicitante las preguntas planteadas por el RMS y el CMS como una Solicitud de Información Complementaria (RSI). Aquí el solicitante dispone de 3 meses (que pueden ampliarse con justificación) para responder. Una vez respondidas, el RMS reiniciará el reloj en el día 106 y actualizará el PrAR y se preparará el FAR.
Evaluación etapa II y discusión del CMD(h)
El RMS reiniciará el reloj en el D106 cuando reciba las respuestas del solicitante a las deficiencias surgidas en los días 70 y 100.
Tendrá lugar una segunda ronda de preguntas por parte de las autoridades: el RMS emitirá nuevos comentarios en el D120 y los CMS, si los tuvieran, en el D145.
Etapa nacional
La autoridad nacional competente de cada Estado miembro implicado adoptará una decisión nacional en un plazo de 30 días después de que el RMS cierre el procedimiento.
El solicitante presentará traducciones nacionales de alta calidad de la ficha técnica, prospecto, el etiquetado y las maquetas en los 5 días siguientes al cierre del procedimiento.
Por lo tanto, podríamos resumir el procedimiento en una línea de tiempo de la siguiente manera:
Conoce la legislación europea
En Qualipharma te ayudamos a definir la estrategia regulatoria más adecuada para poner tu producto en el mercado

